July 31, 2018 at 06:07PM Futurity Weak link could lead to universal flu vaccine
Researchers may have found a weakness in a protein that delivers the flu virus that could be a useful target to stop the virus from infecting cells.
In a new paper, which appears in the Proceedings of the National Academy of Sciences, the researchers delve further into a glycoprotein complex they began to define in a 2014 paper.
That protein, hemagglutinin, sits on the surface of flu viruses and helps them attach to and transport through the protective membranes of target cells.
The paper begins to define the mechanism that allows the protein to unfold and refold in a snap, changing its form to expose a peptide that attaches the virus to a cell and begins infection. The researchers believe therapeutic drugs can use this mechanism to shut the virus down.
Folding and refolding
“This protein starts in a folded state and goes through a global transformation, refolding in a completely different state,” says José Onuchic, codirector of the Center for Theoretical Biological Physics at Rice University. “But there’s a small part in the center that evolution has conserved.”
That single conserved amino acid residue is the hitch that makes the protein pause in the process of refolding. It allows a fusion peptide buried inside to bind to the target cell and begin infecting it. Without the pause, the refolding would be too quick for binding to take place.
Lead author Xingcheng Lin, a postdoctoral research at Rice, modeled that part of the protein, the B-loop of the HA2 domain. HA2 sits beneath another domain, a cap known as HA1 that mutates to escape past defenses. Lin explained that HA1 is a common target for flu medications because the exposed cap domain is more accessible than the protected HA2 domain.
The problem is that HA1 mutates constantly to resist drugs, he says. That influences how effective flu vaccines are every year. Lin and Onuchic says HA2 presents a better target for drugs because the mechanism is highly conserved by evolution.
“If a drug targets HA2, the domain cannot escape by making mutations because the mutations themselves would make it nonfunctional,” Lin says. “That kind of drug could become a universal vaccine.”
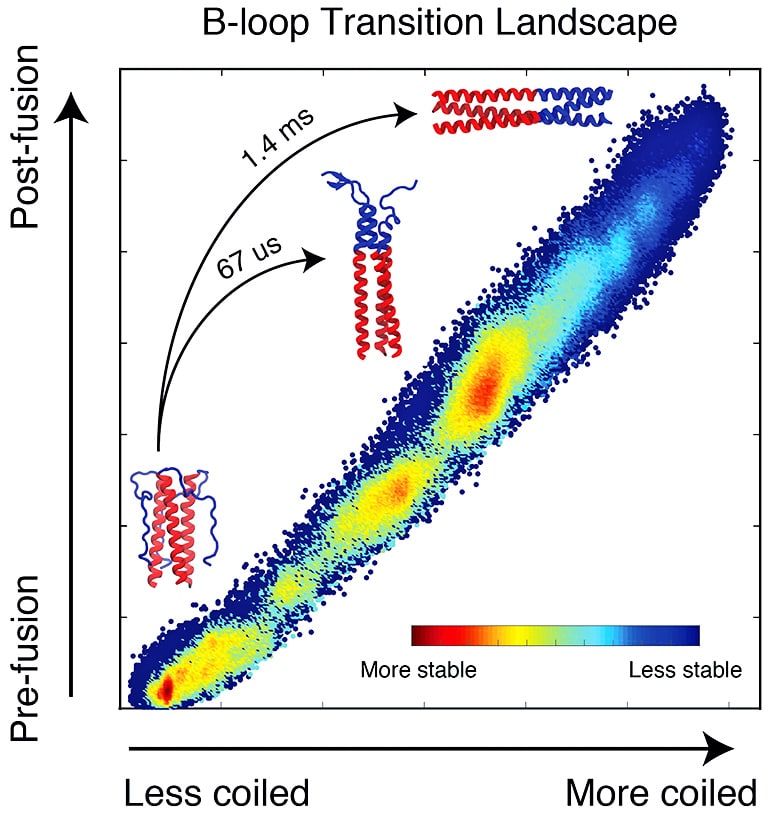
HA2 is a trimeric structure that, when triggered by acidic conditions in the environment near a target cell, transforms itself from a random loop to a coiled coil. Even with the pause, it unfolds and refolds in a fraction of a second, far too fast for microscopes to see. But a computer simulation of the process can be slowed down.
Onuchic and his colleagues are pioneers in the theory that folding proteins follow an orderly, “funneled” process that depends on the intrinsic energy of every atom in the chain, each of which constantly seeks its lowest energy state. If all the atomic “beads” can be identified, it’s possible to simulate the complex folding process.
The researchers often use coarse-grained models of proteins, a subset of atoms that represent the whole, to predict how they will fold. The new study was much more ambitious and set out to predict the complex unfolding and refolding by using not only every atom in the chain but also every atom in its liquid environment, Onuchic says.
‘A gigantic effort’
Lin modeled 40 microseconds (millionths of a second) of the HA2 domain transition that represents the entire process, which takes 1.4 milliseconds (thousandths of a second) to complete. Even that shortened process took two years of computer time to deliver results, he says.
“The simulated domain is about 3,000 atoms, but when the environment, including water, is accounted for, the total simulation incorporates around 100,000 atoms,” Onuchic says. “It’s still an enormous simulation that required state-of-the-art techniques.”
Previous theories based on crystallographic images of the before-and-after proteins put forth the idea of a spring-loaded domain that appeared to attach to the target cell after the cap’s removal. Onuchic says the complete model of HA2 supports a different mechanism.
“We figured out there’s a bunch of energy that makes the final state of HA2 much more stable than the initial state,” he says. “But with the spring-loaded mechanism, most of the energy would already be wasted by the time it forms the coiled coil and binds the cell and viral membranes. It wouldn’t leave any energy to pull the membranes together.
“That’s why we decided to do a full calculation of the system—all the atoms of the protein and all the water,” Onuchic says. “It was a gigantic effort.”
New flu shot is just for horses—but it’ll help us, too
The conserved hydrophilic (water-attracting) residue, known as Thr59, is of particular interest to the researchers not only for the way it disrupts folding and allows the virus to attack, but also because it has a twin.
“In the full evolutionary tree, these viruses fall into two groups, and the difference appears to be this residue,” Onuchic says. “They split 1,500 years ago and somehow, after this separation, they’re fully conserved. They haven’t been able to change that residue no matter what, and we believe that makes this residue important.”
The current research focused on the group that incorporates Thr59 and causes the H3N2 strain responsible for the Hong Kong flu, Lin says. The other residue, Met59, appears in the H1N1 strain that caused the Spanish flu.
“We still have a long way to go to understand the entire protein,” he says. “Here, we only studied one domain of one protein, and there are several others that are very important to its function.”
“But what Xingcheng has already done is a computational tour de force,” Onuchic adds. “He showed how this particular residue breaks the helical symmetry of the domain and makes it unstable enough to give the peptide time to grab the membranes.”
Why men may recover from the flu faster
Additional coauthors are from Rice, Baylor University; and the Max Delbrück Center, Berlin.
The National Science Foundation, the Welch Foundation, and the National Institutes of Health supported the research. The NSF-supported DAVinCI supercomputer, the BlueBioU supercomputer, and the NOTS cluster administered by the Center for Research Computing and procured in partnership with Rice’s Ken Kennedy Institute for Information Technology provided computing resources.
The researchers also used the Anton supercomputer at the Pittsburgh Supercomputing Center, which D.E. Shaw Research made available, as well as the NSF-supported Extreme Science and Engineering Discovery Environment supercomputer.
Source: Rice University
The post Weak link could lead to universal flu vaccine appeared first on Futurity.